Newest
Topics:
For the latest news, see the NEWEST TOPICS page.
Google is too dumb to let me put the list of news in this column and falsely claims that all my pages are self-duplicates.
Google-NONSENSE
Google's so-called "Artificial Intelligence" is an abuse of the concept of intelligence!
A proposed mechanism for the generation of H2
from H2O
in the electron impact ioniser of Mass
Spectrometers
To explain the erroneous H2 analyses
of aqueous Fluid Inclusions
Kingsley Burlinson April 2013
Many researchers have analysed fluid inclusion fluids using
quadrupole mass spectrometers (MS) to identify the chemical
species contained. Some or these researchers have reported the
presence of hydrogen in these fluids. But as discussed in another page on this website, such
analyses are probably incorrect. Laser Raman analyses of fluid
inclusion fluids never find any hydrogen present in these fluids.
The problem is that spurious hydrogen is generated in the ioniser
of mass spectrometers from water and all other hydrogenous species
and this contamination is being ignored. It is assumed that all
the ioniser-produced hydrogen is single atoms of mass 1, which do
not interfere with measurement of hydrogen molecules at mass 2.
But this assumption is unsafe and many spectra of moist air show
the presence of hydrogen at mass 2 (example
shown here). This hydrogen must be generated within the
ioniser but there is no discussion of a mechanism for this.
This discussion suggests a mechanism to explain the origin of
spurious hydrogen of mass 2 from the ionisation of water, in the
electron impact ioniser of mass spectrometers.
The ionisation of water produces copious quantities of H*
and H+ with mass 1. (I use the superscript *
to indicate a neutral free radical atom) It is assumed that
because the mean free path of particles in the ultra-high vacuum
of the mass spectrometer is very long, these particles have no
chance to interact to form H2 with mass 2.
Consequently, any H2 (with mass 2) is assumed to have
been present in the original analyte as hydrogen gas molecules,
rather than being generated in the ionizer. But these assumptions
fail to recognize that fluid inclusion analyses introduce large
amounts or water into the spectrometer vacuum, which greatly
affects the instrument operation.
1. The MS vacuum chamber walls and electrodes are
all coated with copious amounts of water.
Water is a serious problem in ultra-high vacuum systems as it is
"sticky" and adheres to all the chamber walls and prevents
attainment of high vacuum. To get a good vacuum, operators resort
to baking their chambers at some 200 C for hours to desorb water
after any opening of the chamber to change samples! The water
adheres to the chamber walls in a layer several molecules thick.
This quote is from Kurt. J. Lesker Co, who make residual gas
analysers for ultra-high vacuum equipment.
Because fluid inclusion volatiles are almost always dominated by
water, their analyses are done in a "wet" vacuum, in which all the
chamber surfaces are coated with water, which is being continually
introduced during the analyses. The adhesion of water to surfaces
prevents its removal by the vacuum pumps.
2. Ionisation of water produces copious amounts of
H+.
Electron impact ionisation of water produces H2O+
, which is unstable and can then fragment to produce OH+
and H, or alternatively OH and H+ as either fragment
might claim the positive charge. King & Price (Electron
ionization of H2O, Simon J. King, Stephen D. Price,
International Journal of Mass Spectrometry, 277 (2008) 84–90)
document the partial ionisation coefficients for the ionisation of
water and show that the coefficient for H+ is 0.240
(relative to the coefficient of formation of H2O+)
at an ioniser operating voltage of 100 volts. H+ is
also produced during ionisation of all other hydrogenous species
in the analyte, such as hydrocarbons and H2S. There is
an abundance of hydrogen ions of mass 1. What happens to all this
H+? (The table of partial ionisation coefficients
from King & Price is in the appendix, below)
3. H+ hits the
walls of the chamber and interacts with the adsorbed water.
The H+ is attracted by the electric field towards the
entrance aperture of the mass spectrometer, as is the case for all
positive ions. But first there are several focusing and
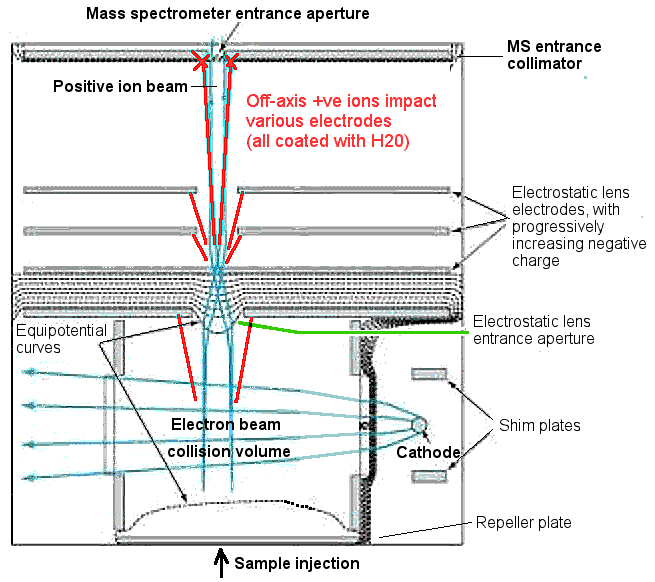
collimation stages for the ions to traverse. This cross section of
an electron impact ioniser shows 5 focusing stages before entry to
the mass spectrometer path. Many ions will hit the focusing and
collimation electrodes and not enter the spectrometer path at all.
If this were a dry vacuum, the ions would merely stick to the
surfaces and cause no trouble. But remember (from discussion
point #1 above), this is a wet vacuum system and all the
focusing electrodes and chamber surfaces are coated with water
molecules. So the H+ is actually impacting a target of
water molecules coating all the electrode surfaces. This is a
BIGproblem!
This is a diagram of a typical electron impact ioniser. Note the
abundant ion impacts (shown in red) with electrodes during
focusing and collimation. Mass spectrometer manufacturers estimate
that only 1 to 3% of the positive ions generated in the
ioniser enter the mass spectrometer aperture. The
overwhelming majority of ions are "off-axis" and impact the
apparatus electrodes and walls, which are all coated with water
molecules.
(image
modified from http://www.defensetechbriefs.com )
4. Hypothesis: H+ interacts with water
adhering to the electrodes to generate H3O, then H2
.
When the H+ ions impact the water molecules adhering
to the electrode surfaces, there is a strong possibility that
various mechanisms could lead to the formation of H2.
One possibility is that it could form hydronium, H3O
and then decompose generating H2. The hydronium
ion is common in interstellar space (an environment quite
similar to a mass spectrometer vacuum!) and decomposes to give OH
and H2. Subsequent H+ impacts could convert
the OH back to H2O. The H2 generated is not
strongly adsorbed on the electrodes and would return to the vacuum
where it could be ionised into H2+ (mass 2)
and then wrongly assumed to have been an original component of the
analyte, when it is in fact spurious contamination. This H2
would have to travel back into the electron beam to be ionised and
this step is problematical, but not impossible and would depend on
the mechanical configuration of the ioniser. Over the long time
frame of fluid inclusion analyses, which can be several hours, the
H2 level in the vacuum could become quite substantial
as it is only poorly removed by the turbo-molecular vacuum pumps
commonly used. The pumping efficiency of these pumps varies
exponentially with the square root of the molecular weight, so
heavier molecules than H2 would be extracted
preferentially, leading to a gradual increase in concentration of
the H2 during the analysis.
Conclusions
The assumption that ionisation of water in the electron
impact ioniser produces only H* or H+
of mass 1 is unsafe as it ignores the very real problem of the
presence of copious quantities of water in the analyte. The
water adheres to all the walls and electrodes in the vacuum
chamber. These aqueous coated and electrically charged
surfaces attract H+ ions whereupon interactions can
occur, probably resulting in H2. Because the
interactions do not occur in the gas phase but on electrode
surfaces, the very long mean free path of particles in the
ultra-high vacuum is irrelevant. The target water molecules
are conveniently attached to all the electrode surfaces
awaiting the inevitable impact of the abundant H+
ions, a perfect configuration to produce complex fragments
including H2.
It is wrong to assume that the ionisation of water only
produces hydrogen atoms or ions of mass 1. Although the
electron impact ioniser appears to be simple, the chemistry
and interactions of all the ion and free radical fragments it
produces is far from simple and must not be ignored as has
been done too often.
H2 (mass = 2) is a common by-product from the
ionisation of water by the electron impact ioniser of mass
spectrometers. This analytical technique cannot be used to
determine hydrogen in aqueous analytes because of the
generation of spurious H2 by the instrument.
Table of coefficients from the study by King
& Price.
Relative partial ionisation cross-sections following electron
ionisation of H2O, expressed relative to the
cross-section for forming H2O+ , as a
function of electron energy E.
E (eV)
µ [H+]
102 *µ [H2+
]
102 * µ [O++ ]
µ [O+ ]
µ [OH+ ]
200
0.261 (11)
0.118 (6)
0.173 (30)
0.0671 (18)
0.315 (3)
175
0.263 (9)
0.119 (18)
0.149 (20)
0.0667 (14)
0.315 (1)
150
0.261 (12)
0.117 (10)
0.109 (11)
0.0651 (18)
0.313 (3)
125
0.255 (12)
0.116 (11)
0.067 (17)
0.0615 (15)
0.310 (2)
100
0.240 (11)
0.113 (8)
0.024 (3)
0.0540 (13)
0.305 (3)
85
0.225 (12)
0.112 (5)
0.005 (3)
0.0466 (17)
0.299 (2)
75
0.209 (9)
0.113 (12)
0.000 (3)
0.0402 (15)
0.293 (3)
65
0.185 (8)
0.109 (8)
0.001 (1)
0.0328 (11)
0.285 (2)
60
0.174 (8)
0.109 (16)
0.000 (1)
0.0292 (18)
0.279 (2)
55
0.160 (8)
0.107 (12)
0.000 (1)
0.0255 (16)
0.273 (2)
50
0.141 (8)
0.107 (12)
0.000 (1)
0.0202 (11)
0.262 (2)
45
0.124 (7)
0.109 (8)
0.000 (1)
0.0159 (10)
0.252 (4)
40
0.109 (6)
0.104 (11)
0.000 (1)
0.0101 (25)
0.239 (5)
35
0.089 (6)
0.096 (12)
0.000 (1)
0.0052 (8)
0.218 (3)
30
0.068 (5)
0.076 (5)
0.000 (1)
0.0013 (9)
0.184 (4)
The value in parenthesis indicates two standard deviations in the
last figure. µ[X+] are the relative partial
ion cross-sections.
NOTE: These coefficients were determined on a "time of flight"
mass spectrometer with conditions such that on average much less
than one ionisation event occurs per pulse of
ionising electrons.
 Applied Mineral Exploration
Applied Mineral Exploration